Exploring the ‘why, how and what’ of this core analytical technique for oral solid dosage forms.
Whether your goal is rapid action or controlled release, from early-stage development through to product quality control (QC), if you’re involved with oral solid dosage (OSD) forms then you’ll be reliant on dissolution testing and robust dissolution test methods. Dissolution tests are highly relevant for other dosage forms, notably semisolids and transdermals and increasingly orally inhaled and nasal drug products (OINDPs). Today though our focus is OSDs.
In vivo, the disintegration and dissolution test of a tablet or capsule is the first step towards therapeutic effect, and control is essential. For many formulators, tablet dissolution testing is therefore a core part of development and quality assurance. Dissolution testing provides critical information to support the realisation of drug release goals, for comparing the performance of different drug substances, for bioequivalence (BE) testing and for product QC. In an earlier blog we talked generally about the tests associated with measuring the critical quality attributes (CQAs) of tablets, today it’s all about dissolution tests and how dissolution testing supports tablet development and QC.
Why do we carry out dissolution testing?
Being clear on the motivation for dissolution testing ensures the firmest foundations for dissolution test method development. So, let’s take a closer look at its relevance and criticality.
When it comes to studying how the body interacts with administered drugs – pharmacokinetics (PK) – the key processes are Absorption, Distribution, Metabolism and Excretion (ADME). These define how the drug moves into, through and out of the body and the drug concentration that establishes in vivo, which is controlled to achieve clinical efficacy and avoid toxicity. For OSDs, disintegration and dissolution are the first, driving steps in this mechanistic chain since absorption is reliant on release of the drug from its delivered form and subsequent solvation. In this way dissolution behaviour influences bioavailability, the ability of the drug to have a therapeutic effect, and by extension the risk of toxicity. Measuring dissolution behaviour is therefore one of the easiest ways to gain valuable insight into drug performance, tablet dissolution behaviour, the consistency of that performance, and safety.
That said, it is important to recognise that absorption is not governed solely by dissolution. Permeability through the cell membranes of, typically, the gastrointestinal tract is equally important, as reflected in the Biopharmaceutics Classification System (BCS) which groups drugs on the basis of solubility and permeability, in combination (see below).
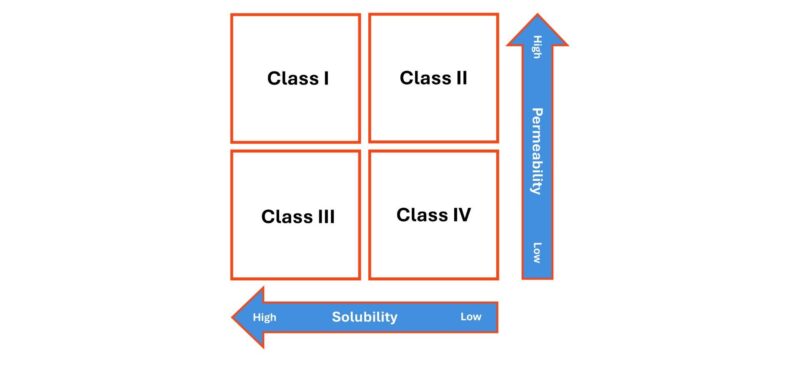
Class I and II drugs have high permeability and will therefore pass relatively easily into circulation once dissolved. For Class II drugs, which currently dominate the drug development pipeline this means bioavailability is likely to be dissolution rate limited. BCS class II drugs have been a focus for solubility enhancement research in recent times, and several formulation approaches for this class of compound have been developed.
Class III and IV drugs, in contrast, will not absorb easily, even when dissolved, due to poor permeability. For these drugs there is potential for drug concentrations to become relatively high on the ‘delivery’ side of the membrane while remaining very low in the bloodstream.
Also, note the use of the term solubility on the bottom axis. When it comes to dissolution behaviour, we can investigate both the speed of dissolution and the extent to which the drug is soluble in different media. Both are important.
How do we use dissolution test data?
Data obtained from dissolution studies and dissolution test programmes drive choices and progress throughout formulation helping to differentiate APIs (active pharmaceutical ingredients), excipients, formulations, and manufacturing strategies on the basis of their ability to enhance bioavailability. Such data also support increasingly sophisticated PK modelling to efficiently accelerate drugs to market and help to safeguard quality across the entire lifetime of a drug, through generic development and indeed any transition to over-the-counter provision.
Let’s examine the use of dissolution data in more detail by considering questions that they can help to answer which include:
- Which form of the drug is preferable for a rapid onset product?
- Is there scope to improve dissolution characteristics by changing the manufacturing route of the API, to control properties such as particle size or shape?
- Can I identify excipients that enhance the rate of dissolution or increase solubility? Is all the drug released during the dissolution process?
- Which excipients are most advantageous in terms of dissolution behaviour? And which are detrimental?
- Will this coating or matrix enable sustained drug release? Is the drug release profile optimal for clinical efficacy?
- How do tableting conditions such as compaction pressure, and tablet properties such as hardness, impact tablet dissolution behaviour?
- Has the change in API or excipient supply impacted the dissolution profile?
- Does this test product deliver the same dissolution profile as the reference?
- Is this batch of tablets equivalent to the batch made last week, last month, ten years ago when assessed through tablet dissolution testing?
These questions illustrate the breadth of application of dissolution test data and the need to think carefully about test methods to optimise their ability to address different issues.
What factors inform dissolution testing methods?
USP Apparatus 1 (Basket – left) and Apparatus 2 (Paddle – right) are the most common dissolution test set-ups, and are widely used in modern dissolution testers. In future blogs we’re going to look at the test methods associated with these and the other five compendial apparatuses for dissolution testing in more detail. Here though we wanted to highlight some broader points associated with test methods.

Firstly, it’s clear that there are decisions to be made over the dissolution media used, whether to simulate the fed or fasted state, for example, whether to modify pH or other aspects of the dissolution media to reflect dissolution in different areas of the gastrointestinal tract, and/or the appropriate level of agitation for representative testing.
Secondly, there is the issue of localised drug concentrations. The relatively large volume of dissolution test apparatus means that testing tends to be carried out under ‘sink’ conditions, in other words under conditions that maintain the localised drug concentration at such a low level that there is no impact on dissolution behaviour. This may be the preference. But, if, in practice the drug will necessarily dissolve into a relatively concentrated solution, because the goal is extended release, for example, or because of poor permeability (Class 3 and 4 drugs), then alternative strategies may arguably be more relevant.
Thirdly, the growing number of Class II drug candidates intensifies the need for sensitive dissolution testing. Large test volumes in combination with sparingly soluble drugs translate into a requirement to measure drugs at very low concentrations, challenging assay methods. Reducing test volumes may alleviate this problem, and there are off-the-shelf solutions to do this, but more generally such testing calls for enhanced precision with respect to both apparatus and method.
Last, but not least, the broad applicability and value of dissolution test data highlights the importance of test set-ups and methods that are highly productive. Investing in equipment that makes dissolution as accurate, efficient, and streamlined as possible is therefore extremely worthwhile.
In conclusion
Dissolution test methods are well-established but continue to evolve in line with requirements for tablet dissolution testing, challenging drug candidates, and increasingly sophisticated OSD products. Being clear on the purpose of testing is vital to ensure test set-ups and methods are as relevant as possible. If you’re interested in learning more about the basics of dissolution testing, then we have a great introductory article; alternatively, look out for future blogs when we’ll be looking at the practicalities of testing in more detail.








